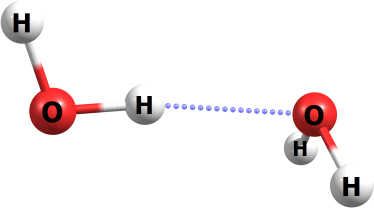
Water Dimer
Characterize the binding energy of the water dimer
This exercise will show you how to determine the binding energy for the water dimer in the non-planar open Cs conformation1.
Overview
The binding energy (Ebind) of two interacting molecules can be determined using the following equation:
\[ E_{\mathrm{bind}} = E_{\mathrm{dimer}} - \left ( 2 \times E_{\mathrm{monomer}} \right ) \]
where Edimer is the energy of the dimer and Emonomer is the energy of the monomer. Because two water molecules favorably interact, there is a lowering of the potential energy. By subtracting the energy of two, non-interacting optimized water molecules from the energy of optimized dimer system, this gives the energy of the interaction (i.e. the binding energy).
Therefore, we must optmize a single, isolated water molecule as well as the dimer complex. We will examine the low-energy non-planar open Cs conformer of the water dimer (pictured below).
Setup and Run
Create the following directories.
~/chem/water/monomer/b3lyp/dz/~/chem/water/dimer/b3lyp/dz
Within each directory, create input files, one for the water monomer and one for the water dimer. Be sure to include a copy of the PBS file in each directory.
WATCH: Setup the directory and input file for the water monomer.

Commands:
mkdir -p ~/chem/water/monomer/b3lyp/dzto make the directory from yourHOMEdirectorycd ~/chem/water/monomer/b3lyp/dzto change into the directoryvi 1.inpto create the input fileito enter Insert Moderight click(ormiddle click) to pasteEnterto insert a blank line at the end of the fileESCto enter Command Mode:wqto save and close the file
You will notice that the molecule specification in the input file is given as a Z-matrix and not as Cartesian coordinates. The Z-matrix is carefully crafted to ensure the C2v symmetry of the monomer and the Cs symmetry of the dimer.
Submit the job to the queue.
% qsub g09.pbsYour job should now be in the queue. To check if it has been queued up, type
% qstat | grep $USERThis will only show your jobs that are in the queue. Once the job completes, it will no longer be in the queue. A 1.out file should be in your directory if the job ran.
Analyze the Output File
Water Monomer
The output file contains a plethora of information and a primer on interpreting the output file is covered here. For now, we want to pull the final electronic energy from the output file.
% grep "SCF Done:" 1.outWATCH: Pull the electronic energies from the output file.

Commands:
grep "SCF Done:" 1.outwill print to the screen every line in1.outthat contains the string “SCF Done:”.
The last energy is what we want. Another command to type to simply get the last energy is
% grep "SCF Done:" 1.out | tail -1
SCF Done: E(RB3LYP) = -76.4206270785 A.U. after 1 cyclesYou must also check the nature of the stationary point from the frequency computation. The optimized geometry should be a minimum on the potential energy surface. Verify this by ensuring all the vibrational modes are real (not printed with a negative sign).
% grep "Frequencies" 1.out
Frequencies -- 1658.8679 3751.3292 3852.7337WATCH: Pull the frequencies from the output file.

Commands:
grep "Frequencies" 1.outwill print to the screen every line in1.outthat contains the string “Frequencies”.
Here, all the frequencies are real (reported as positive numbers). Therefore, the geometry is indeed a minimum (on the B3LYP/cc-pVDZ potential energy surface).
Water Dimer
Do the same with your water dimer output file.
% grep "SCF Done:" 1.out | tail -1
SCF Done: E(RB3LYP) = -152.854436576 A.U. after 1 cyclesWe now have our energies to calculate the binding energy.
Binding Energy
Let us convert our absolute electronic energies into binding energies according to the equation at the beginning of this tutorial. I will then convert this energy to kcal mol–1 by multiplying by 627.51. This can be done in an Excel spreadsheet for convenience.
Here we see that the binding energy for our water dimer is –8.27 kcal mol–1 on the B3LYP/cc-pVDZ potential energy surface. The binding energy is negative indicating an attractive interaction.
Other Binding Energies
An experimental value for the binding energy of the water dimer is –5.4±0.7 kcal mol–1.2
Below are computed binding energies (in kcal mol–1) of the water dimer as fully characterized at the indicated methods and basis sets.
Below is a plot of the data in the table above. The solid black line represents the experimental value (solid black line) and the experimental error (dashed black lines) are included.
Questions
- Which levels of theory agree well with experiment?
- What levels of theory would you not recommending using?
- What other factors should you consider when comparing these results to experiment?
- Would you expect these computations to agree well with experiment? How can one make better predictions?
Suggested Reading
- Gillan, M. J.; Alfè, D.; Michaelides, A. Perspective: How Good Is Dft for Water? J. Chem. Phys. 2016, 144 (13), 130901. https://doi.org/10.1063/1.4944633.
References
(1)
Tschumper, G. S.; Leininger, M. L.; Hoffman, B. C.; Valeev, E. F.; Schaefer, H. F.; Quack, M. Anchoring the Water Dimer Potential Energy Surface with Explicitly Correlated Computations and Focal Point Analyses. J. Chem. Phys. 2002, 116 (2), 690–701. https://doi.org/10.1063/1.1408302.
(2)
Curtiss, L. A.; Frurip, D. J.; Blander, M. Studies of Molecular Association in H2O and D2O Vapors by Measurement of Thermal Conductivity. J. Chem. Phys. 1979, 71 (6), 2703–2711. https://doi.org/10.1063/1.438628.